
Spatial Transcriptomics and scRNA-seq: A Complementary Pair
The current state of spatial transcriptomics and scRNA-seq: single-cell-resolution platforms, foundation models for integration, spatial multi-omics, and 3D tissue atlases.
Single-cell and spatial transcriptomics series, Part 3 of 3. Part 1 covers when scRNA-seq alone is enough and Part 2 covers integrating spatial data. This final part looks at where the two technologies now stand and where they are heading.
Part 1 looked at what scRNA-seq answers on its own, from cataloguing cell types and states to differential expression and trajectories. Part 2 added spatial context and the methods for integrating the two. This part is about the current state of the field and the near future: the spatial platforms that now reach single-cell resolution, the machine-learning and foundation models doing the integration, spatial multi-omics that go beyond RNA, and the move to 3D tissue atlases. The through-line is that scRNA-seq and spatial transcriptomics are no longer an either-or choice; increasingly a study uses both, with single-cell data serving as the deep reference that gives spatial data its cell identities.
The current spatial platforms
Spatial transcriptomics has moved quickly. Until recently the field split cleanly into unbiased-but-coarse (spot-based sequencing) and precise-but-targeted (imaging). That split has largely collapsed. Sequencing-based platforms now reach single-cell scale, and imaging platforms have pushed from hundreds of genes to thousands, or the whole transcriptome. The current landscape:
| Platform | Type | Resolution | Plex (as of 2026) | Notes |
|---|---|---|---|---|
| Visium HD (10x) | Sequencing capture | 2 µm, single-cell scale | Whole transcriptome (probe-based) | FFPE-compatible; standard since 2024 |
| Xenium / Xenium Prime (10x) | Imaging | Subcellular | Up to 5,000 genes (Prime 5K) | Protein co-detection; large sections |
| CosMx SMI (Bruker) | Imaging | Single-cell to subcellular | 6,000-plex, or ~19,000 (whole transcriptome) | RNA and protein together |
| MERSCOPE / MERFISH (Vizgen) | Imaging | Subcellular | ~500 to 1,000 genes | High per-transcript sensitivity on targeted panels |
| Stereo-seq (STOmics) | Sequencing (DNB array) | Nanoscale, subcellular | Whole transcriptome | Very large capture areas, whole embryos or organs |
| Slide-seqV2 / Curio (bead) | Sequencing | ~10 µm, near single-cell | Whole transcriptome | Bead-array capture |
Two lines have converged here. On the sequencing side, Visium HD replaced the old 55 µm spots with a continuous 2 µm array across the capture area, and Stereo-seq reaches subcellular resolution over very large fields. On the imaging side, Xenium Prime now runs 5,000-gene panels and CosMx offers a 6,000-plex panel plus a whole-transcriptome (~19,000 gene) capability, both at single-cell to subcellular resolution. scRNA-seq still provides the deepest and most unbiased per-cell readout, and it remains the reference used to annotate and interpret these spatial datasets.
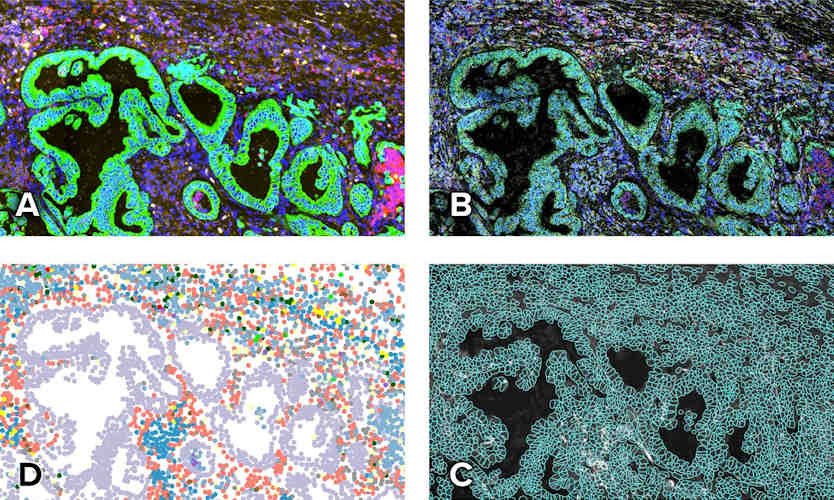
Figure: CosMx (Bruker Spatial Biology) output on human lung cancer tissue, an imaging platform that profiles RNA and protein at single-cell resolution. Immunofluorescence images (top) are used to delineate individual cells and assign cell types (bottom), building a spatial cell map of the tissue in situ.
Single-cell-resolution atlases at scale
As resolution and throughput have risen, spatial data has started to behave like another form of single-cell data, one that carries x,y (and sometimes z) coordinates. That has enabled spatially resolved atlases where every point is a cell with a rich gene profile.
The whole mouse brain atlas from the BICCN consortium is the clearest example: it combined a single-cell RNA-seq dataset of around 7 million cells with a MERFISH spatial dataset of around 4 million cells, and defined roughly 5,300 cell-type clusters, each placed in the brain's anatomy (Yao et al., 2023). Defining what a cell is from its transcriptome and where it sits from spatial data is now a standard atlasing pattern, and the Human Cell Atlas and related efforts are applying it organ by organ. Purpose-built methods keep pushing resolution: Seq-Scope reaches submicron capture, and sci-Space tags nuclei in place before sequencing to produce spatially resolved single-cell transcriptomes at scale.
The practical effect is on experimental design. The question is less often "scRNA-seq or spatial?" and more often "which of each, in what order?" A common pattern is to run scRNA-seq to discover cell types and gene programmes, then a spatial assay to see how those types are arranged and interact, with each dataset informing the other.
Machine learning and foundation models for integration
Combining single-cell and spatial data at this scale is a computational problem, and the methods have kept pace. For mapping single cells onto tissue, cell2location and Tangram remain widely used, and label-transfer and reference-mapping tools (Seurat, scVI, scArches) project annotations from a reference onto new data. For finding spatial structure, graph neural network methods such as SpaGCN and STAGATE define tissue domains by combining expression similarity with physical proximity. Probabilistic frameworks like scVI and totalVI handle batch correction and joint embedding across modalities, including combined RNA and protein.
The newer development is foundation models. Transformer models pretrained on tens of millions of cells, such as scGPT (Cui et al., 2024) and Geneformer (Theodoris et al., 2023), learn general representations of cells and genes that can be fine-tuned for annotation, batch integration, and gene-network or perturbation inference. Work to extend this class of model to spatial context is active, aiming for models that reason jointly over expression and tissue position. These approaches are promising rather than settled; benchmarks show they do not always beat well-tuned conventional methods, so they sit alongside the established tools rather than replacing them.
Spatial multi-omics: beyond RNA
Transcription is one layer. The clearer trend now is measuring several molecular layers in the same tissue, so that gene expression can be read against what is actually executed in the cell and how it is regulated.
| Layer | What it adds | Example spatial methods |
|---|---|---|
| Transcriptome | Which genes are on, per location | Visium HD, Xenium, CosMx, Stereo-seq |
| Proteome | What the cell is actually executing | CODEX / PhenoCycler, MIBI, CosMx and Xenium protein co-detection |
| Epigenome | How expression is regulated | spatial-ATAC-seq, spatial-CUT&Tag, joint epigenome-transcriptome co-profiling |
| Metabolome | Local metabolites and lipids | imaging mass spectrometry (e.g. MALDI) |
Spatial proteomics is the most mature addition. Imaging platforms like CODEX (PhenoCycler) and MIBI stain dozens of proteins in tissue, and CosMx and Xenium already co-detect protein alongside RNA, which matters for reading cell state or drug response in situ (for example immune-checkpoint or phospho-protein markers next to gene expression). NanoString's GeoMx Digital Spatial Profiler, now part of Bruker, uses photocleavable barcodes to read tens to thousands of proteins or RNAs from selected regions.
The epigenome layer is no longer nascent, which is the biggest change from a couple of years ago. Spatial ATAC-seq maps chromatin accessibility across tissue at near-single-cell resolution (Deng et al., 2022), and joint methods now co-profile the epigenome and transcriptome on the same section, reading chromatin accessibility or histone modifications alongside mRNA (Zhang et al., 2023). Spatial metabolomics via imaging mass spectrometry adds metabolite and lipid maps that can be layered onto cell maps to study, for example, nutrient gradients around a tumour. Put together, these layers let you ask integrated questions: at an injury border in heart tissue, which genes are upregulated, which signalling proteins are active, and how is chromatin remodelled, all in spatial register.
Toward 3D tissue atlases
Biology is three-dimensional, but most spatial experiments are run on 2D sections. Reconstructing 3D volumes is an active frontier, either by stitching serial sections computationally or by capturing volumes directly.
Open-ST is a good current example of the sequencing route: an open-source method that captures transcripts at subcellular resolution and reconstructs tissue in 3D from serial sections (Schott et al., 2024). Applied to a head-and-neck tumour and patient-matched metastatic lymph node, the 3D reconstruction revealed spatially contiguous structures and candidate biomarkers at the tumour-lymph interface that were not visible in any single 2D section. On the imaging side, STARmap demonstrated 3D in-situ sequencing directly in intact tissue blocks using hydrogel chemistry.
Large atlas projects are building 3D references at scale. The whole mouse brain atlas is effectively a volumetric map, and the NIH's HuBMAP program is assembling a 3D atlas of the human body at cellular resolution, integrating spatial transcriptomics, imaging, and single-cell sequencing into common coordinate frameworks organ by organ. These atlases are practical tools: a reference to compare a patient's tissue against, and context for interpreting where a cell type normally resides and what it does. In drug discovery, knowing the spatial distribution of cell types can point to which cells to target and where, for instance a niche of fibroblasts near airways driving lung fibrosis.
Where ScarfWeb fits
Handling this range of data needs software that does not assume the user is a bioinformatician. ScarfWeb, Nygen's browser-based single-cell platform, is technology-agnostic: it supports droplet and plate-based scRNA-seq, CITE-seq (RNA and protein), BCR/TCR sequencing, and spatial coordinate import for visualising clusters on tissue layouts. Analyses run in the cloud with no installation, which matters when single-cell matrices and spatial images get large, and results can be shared as interactive views rather than passed around as files. That makes it a workable hub for pulling single-cell and spatial data into one place, which is increasingly what a tissue study requires.
Closing the series
Across three parts the pattern has been consistent: the identity of a cell, from scRNA-seq, and its location and neighbours, from spatial methods, together explain more than either does alone. What has changed recently is how far the tools have come. Spatial platforms now reach single-cell resolution, integration methods and foundation models are maturing, multi-omic layers including the epigenome can be read in spatial register, and 3D reconstructions are moving from proof-of-concept to published biology.
For anyone planning experiments, the takeaway from the series is practical. Decide first what the question actually needs (Part 1), add spatial data where arrangement and interactions matter (Part 2), and expect to use both modalities together as the default rather than the exception. scRNA-seq remains the reference layer underneath all of it.
References
Yao, Z., et al. (2023). A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature, 624, 317-332. https://doi.org/10.1038/s41586-023-06812-z
Cui, H., Wang, C., Maan, H., et al. (2024). scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 21, 1470-1480. https://doi.org/10.1038/s41592-024-02201-0
Deng, Y., et al. (2022). Spatial profiling of chromatin accessibility in mouse and human tissues. Nature, 609, 375-383. https://doi.org/10.1038/s41586-022-05094-1
Zhang, D., et al. (2023). Spatial epigenome-transcriptome co-profiling of mammalian tissues. Nature, 616, 113-122. https://doi.org/10.1038/s41586-023-05795-1
Schott, M., León-Periñán, D., Splendiani, E., et al. (2024). Open-ST: high-resolution spatial transcriptomics in 3D. Cell, 187(15), 3953-3972. https://doi.org/10.1016/j.cell.2024.05.055
Oliveira, M. F., Romero, J. P., Chung, M., et al. (2025). High-definition spatial transcriptomic profiling of immune cell populations in colorectal cancer. Nature Genetics, 57, 1512-1523. https://doi.org/10.1038/s41588-025-02193-3
Meng, F., Aierken, A., Li, F., et al. (2024). Advances in spatial transcriptomics technologies and applications in cancer research. Frontiers in Cell and Developmental Biology, 12, 1378875. https://doi.org/10.3389/fcell.2024.1378875
Hu, J., et al. (2023). Spatial transcriptomics reveals distinct and overlapping functions of cancer-immune microenvironments with impacts on biology and therapy response. Nature Communications, 14, 4526. https://doi.org/10.1038/s41467-023-40271-4
Related articles
The biology in your ligand-receptor analysis, and how to read it
Ligand-receptor analysis is easy to run and easy to over-read. How to get cell-cell communication biology from the ranked table, with LIANA and ScarfWeb.
Read more →
Multi-Omics Data for Target ID in Drug Discovery
How human genetics, single-cell transcriptomics, chromatin accessibility and proteomics combine into causal, cell-resolved target evidence, where integration usually fails, and how to grade the result before a programme is committed.
Read more →
Integrating Spatial Transcriptomics with Single-cell RNA-seq
How to integrate spatial transcriptomics with scRNA-seq: when spatial context helps, the key integration methods, and worked tissue examples.
Read more →