
What We Got Wrong About AI Agents for Cell Annotation
Running CyteType's AI agents across thousands of single-cell RNA-seq datasets in production exposed run-to-run variance, selective evidence gathering, and inconsistent depth. We rebuilt cell type annotation as a deterministic LLM workflow for reproducible, auditable results in drug discovery pipelines.
In June 2025, we published an article titled "Why We Built AI Agents That Actually Understand Single Cell Data." In it, we made the case that cell type annotation required a team of specialized AI agents working in concert, each with autonomy to reason through evidence, query databases, and reconcile conflicting biological signals. We showed how CyteType's multi-agent architecture identified misclassified cell types across 40,000+ cells in a public atlas. We were proud of the work.
We still stand by the core decomposition. Breaking annotation into hypothesis generation, evidence collection, review, and literature validation was the right call. The problem was with how we let those steps execute.
Over the past several months, we've been running CyteType across thousands of datasets in production. What we observed forced us to rethink how AI should be applied to recurring analytical tasks in single-cell biology. We changed direction in January. This post explains what we learned and why.
What the agents were actually doing
In CyteType's original architecture, each agent had a preset role but autonomy within that role. Take the evidence collection step as an example. The agent's job was to evaluate whether a candidate cell type, say CD4+ T cells, was supported by the expression data.
In practice, the agent would decide on its own: which genes to check, how many to examine, in what order, and how to weight them. On one run it might evaluate 5 marker genes. On another, 150. Both runs would produce a confident annotation with a reasoning chain, and both would look perfectly reasonable on inspection.
That variability is the problem.

When an agent checks 5 genes for one cluster and hundreds for another, the thoroughness of the annotation becomes a function of the model's planning step, not a function of the biology.
Three things we observed in production
1. Variance between runs on identical data.
We ran the same dataset through CyteType's agentic pipeline multiple times. The annotations were not always the same. Not because the biology changed, but because the agent's planning step is non-deterministic. It might query databases in a different order, weight supporting evidence differently, or generate a slightly different reasoning chain. For production use, where annotations need to be consistent across studies, time points, and therapeutic programs, that's not acceptable.
2. Selective evidence gathering.
Agents are optimised to produce an answer. When marker gene evidence is ambiguous, particularly for transitional cell states, disease-perturbed populations, or rare cell types, the agent would selectively cite markers that supported its conclusion while underweighting contradictory signals. This isn't a prompting failure. It's an emergent property of goal-directed architectures. The agent is trying to complete the task, and completing the task means arriving at an annotation.
3. Inconsistent depth across clusters.
Some clusters received deep, thorough analysis. Others got a surface-level pass. We had no programmatic way to enforce equal rigour across every cluster in a dataset. We could prompt the agent to be thorough, but we were relying on the model to follow that instruction faithfully every time. In practice, it didn't.
What we changed
Starting in January, we rebuilt CyteType's internal architecture as a deterministic AI workflow. The LLMs are still there. They still provide the contextual reasoning that makes annotation possible. But they no longer decide what to do next.

Here's how the evidence collection step works now, using a simplified example. Given a candidate cell type, we ask the LLM a specific question: "What are N genes whose expression would support this annotation, and why?" where N is a fixed number we define. The LLM returns a structured list with reasoning for each gene. We then programmatically check every one of those genes against the actual expression data. No LLM involvement in that step.
Next, we ask a separate LLM call: "What are N genes whose presence would negate this annotation, and why?" Same process. Programmatic verification against real data.
Then we go back to an LLM with the compiled results: "Here is the expression data for each of your supporting and negating genes. For each gene, based on your original reasoning and the observed data, is this gene supporting or not supporting the candidate?" The LLM evaluates gene by gene, producing a binary score that we aggregate programmatically into a confidence measure.

The difference is control. We control the depth (a fixed number of genes per candidate, every time, not 5 or 500 depending on the model's mood). We control the breadth (always both supporting and negating evidence). We control the reasoning structure (always gene-by-gene evaluation against real expression data). And because each LLM call is small and transactional, we can build in programmatic retries cheaply. If the model returns fewer genes than requested, we can say "you've given me these, give me the rest" and retry at minimal cost.
None of this is possible in an agentic architecture where the model is deciding what to check and when to stop.
What we got right, and what we got wrong
We got the decomposition right. Annotation genuinely requires distinct reasoning phases: hypothesis generation, evidence collection, independent review. That structure remains in CyteType, and it's documented in our preprint.
What we got wrong was giving the LLMs autonomy within those phases. We described CyteType as a "multi-agent system" where agents had the freedom to reason through problems. In reality, that freedom introduced variance, selective evidence gathering, and inconsistent depth, all things that are specifically harmful for a recurring analytical task that needs to produce defensible, auditable results.
The shift can be summarised simply: we went from giving LLMs independence to micromanaging them. Every LLM call now has a specific, bounded question. The workflow controls what happens between calls. The result is that CyteType produces equally thorough analysis for every cluster, every time, on every dataset.
What comes next
We said we would return to a broader question: in an industry racing toward autonomous AI agents for drug discovery, when should you use an agent, and when should you use a workflow? We did, in a follow-up on agents versus workflows in drug discovery. It walks through where agent architectures genuinely fit, where deterministic workflows do, and how we think about that distinction on a computational biology team, including why we rebuilt CyteType around a workflow for one task rather than an open-ended agent.
The original article, "Why We Built AI Agents That Actually Understand Single Cell Data," remains live on our blog. It represents where our thinking was at the time, and we think there's value in showing the full trajectory of how a product evolves when you actually run it in production.
CyteType is available via pip install cytetype (Python) and from GitHub (R via CyteTypeR). If you're evaluating AI-based annotation tools for your single-cell pipeline, we'd welcome the conversation. Schedule a demo →
Related articles

Cherry-picking is a bigger annotation risk than hallucination
Cherry-picking in LLM cell annotation survives reference checks unlike hallucination. Learn why biased marker subsampling is the harder risk and how CyteType prevents it.
Read more →
Everyone Is Building AI Agents for Drug Discovery. We Built a Workflow for One Task. Here's Why.
Why we rebuilt CyteType as a deterministic AI workflow instead of an agent, and why that distinction matters for production cell annotation pipelines.
Read more →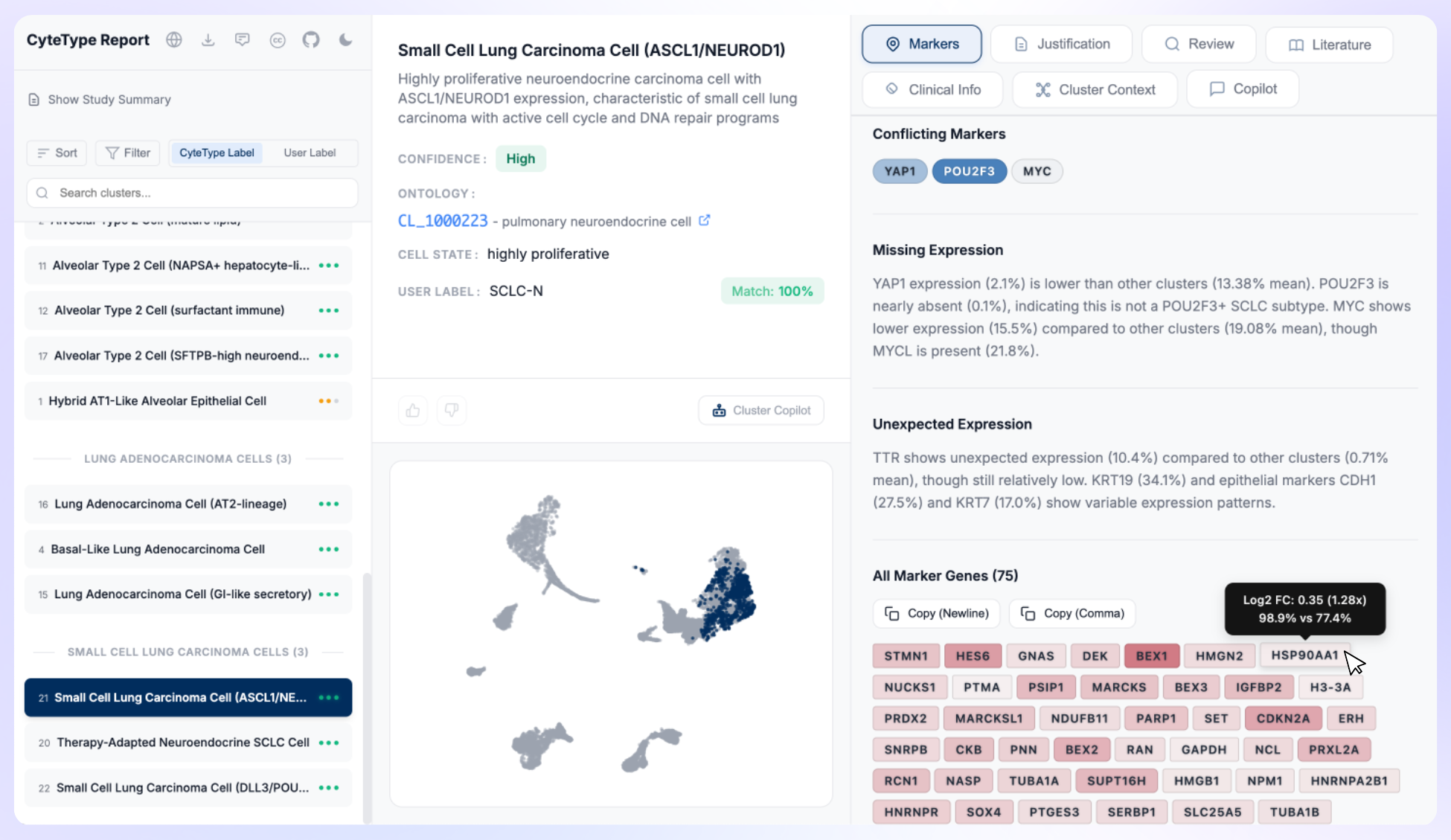
CyteType: Evidence-Based Cell Annotation with Multi-Agent AI
CyteType uses a five-agent AI framework for accurate cell type annotation in scRNA-seq data. Outperforms reference-based methods by 300%+ in benchmarking.
Read more →