
Multi-Omics Data for Target ID in Drug Discovery
How human genetics, single-cell transcriptomics, chromatin accessibility and proteomics combine into causal, cell-resolved target evidence, where integration usually fails, and how to grade the result before a programme is committed.
Target attrition is an evidence problem before it is a chemistry problem
Around nine in ten candidates entering clinical development never reach approval, and the largest single contributor is insufficient efficacy in Phase II. That is usually not a formulation problem or a potency problem. It is a target problem: committing to a mechanism whose causal relationship to human disease was assumed rather than demonstrated.
Human genetic support remains the durability benchmark. Targets with genetic evidence linking the gene to the indication progress through clinical development at roughly twice the rate of those without it, a result first quantified by Nelson and colleagues in 2015 and reproduced by King and colleagues in 2019. Every other molecular layer should be judged by the same standard: does it move the candidate on one of these axes?
-
01
Causality
Human genetics, colocalisation, or Mendelian randomisation that support the therapeutic hypothesis.
-
02
Cellular context
Which cell type or state mediates the effect in diseased human tissue.
-
03
Direction of effect
Whether perturbing the target shifts the disease programme the expected way.
Assembling more modalities without moving these axes is not progress.
What each layer can and cannot answer
The practical value of an integrated analysis depends on knowing where each assay's inference stops.
| Layer | What it can establish | What it cannot settle alone |
|---|---|---|
| Human genetics (GWAS, rare variant burden, Mendelian randomisation, colocalisation) | Causal direction from germline variation to disease; loss-of-function tolerance as a first safety read | Which cell type or state mediates the effect; whether the effect is developmental or ongoing |
| Epigenomics (scATAC, 10x Multiome, promoter capture Hi-C) | Assignment of non-coding risk variants to candidate effector genes and to the regulatory programmes active in a given compartment | Whether the regulatory change propagates to protein and phenotype |
| Transcriptomics (single-cell, single-nucleus, spatial) | Cell state, compartment, heterogeneity, and composition shifts that accompany disease | Activity. Transcript abundance is a poor proxy for functional protein in many cell types |
| Proteomics (affinity plasma proteomics, pQTL, single-cell mass spectrometry) | Effector-level abundance and, through pQTL-anchored PheWAS, an early view of on-target liabilities | Localisation and post-translational state; affinity panels are vulnerable to epitope-disrupting variants |
| Metabolomics | Pathway flux constraints and metabolic dependencies with phenotypic consequence | Cell of origin, which is almost always inferred indirectly |
Convergence is the signal worth acting on. A gene under a fine-mapped locus, with accessibility and expression restricted to the disease compartment, a plasma pQTL consistent in direction with the association, and a perturbation that moves the relevant programme, is a different proposition from a gene that is merely differentially expressed in bulk tissue.
Targets live in cell states, not tissues
Regulatory genetics has become cell-resolved. Single-cell eQTL studies, notably the OneK1K cohort with roughly a thousand donors profiled by scRNA-seq, showed that a substantial fraction of regulatory effects are restricted to specific cell types and, in some cases, to activation states within those types. Colocalisation against bulk tissue eQTL will miss those effects, or assign them to the wrong effector gene when the dominant cell type is not the disease-relevant one.
The corresponding biology has moved the same way. The population that matters therapeutically is often a state, not a coarse lineage label:
- Fibrotic lung disease: SPP1+/TREM2+ profibrotic macrophages (Adams and colleagues; replicated across IPF cohorts), not "macrophage"
- Neurodegeneration: disease-associated microglia (Keren-Shaul and colleagues), later fractionated into stress-primed and terminal states with different disease specificity
- Solid tumours: FAP+ matrix-remodelling fibroblasts and LAMP3+ mature regulatory dendritic cells, neither of which survives a coarse stromal or myeloid label
Cell-type-resolved colocalisation, compartment-specific differential expression, ligand-receptor inference, and composition analysis all take the cell annotation as a fixed input. It is an estimate. Every downstream inference inherits its error, and there is rarely a diagnostic that surfaces the problem before the target list is written.
Where integration usually breaks
Four failure modes account for most analyses that look convincing and do not replicate.
Design rather than biology dominates the variance. Modalities are seldom collected on the same cells, often not on the same samples, and sometimes not on the same cohort. Disease status is frequently confounded with site or protocol. A single donor dominating a disease-enriched cluster is common in small cohorts and invisible in a UMAP.
Pseudoreplication inflates differential expression. Treating cells as independent replicates when the biological unit is the donor produces false positives (Squair and colleagues, 2021). Pseudobulk within donor and cell type remains the defensible default; regenerate any target list built on cell-level testing.
Joint embeddings can be corrected past usefulness. Methods that remove batch structure also remove condition structure when the two are correlated. The result can look better on integration metrics and worse on biology. Read batch and conservation metrics together.
Annotation is treated as settled when it is not. Coarse labels hide the state that carries the mechanism; confidently wrong labels are worse because they close off inspection. Cell-type-specific evidence for a candidate may be an artefact of how clusters were named.
Matching method to design
Method choice should follow from the experimental design and the question, not from what performed well in the most recent benchmark.
| Study design / question | Prefer |
|---|---|
| Cohort bulk or pseudobulk layers | MOFA+ (MEFISTO if temporal or spatial structure) |
| Paired RNA + protein (CITE-seq) | Weighted nearest neighbours or totalVI |
| Paired or partially paired RNA + ATAC | MultiVI |
| Unpaired modalities | GLUE-class graph linking (state the linking assumptions) |
| Signalling or tissue architecture | LIANA; tensor-cell2cell across conditions or time |
None of this establishes direction of effect. Perturbation does. Genome-scale Perturb-seq, targeted CRISPR screens in disease-relevant primary cells, and functional proteomic readouts convert convergent association into a testable claim. Send the shortlist there quickly rather than accumulating further observational layers.
Grading the evidence before it becomes a programme
A target nomination is easier to defend, and easier to kill, when the evidence is scored explicitly rather than described narratively.
- Causal support. Is there human genetic evidence, and does Mendelian randomisation or colocalisation support the direction assumed by the therapeutic hypothesis?
- Cell context. Is activity localised to a defined cell type or state present in human diseased tissue at a frequency that matters?
- Direction of effect. Does perturbation shift the disease programme the expected way in a relevant model?
- Tractability and early safety. Modality fit, normal-tissue expression, constraint metrics, and pQTL-anchored phenotypic associations that hint at on-target liability.
- Translational bridge. Does the state targeted in the preclinical model correspond to a state that exists in patient tissue?
Where a candidate scores well on convergence but the underlying cell annotations have not been examined, criterion two is unresolved regardless of how strong the rest looks.
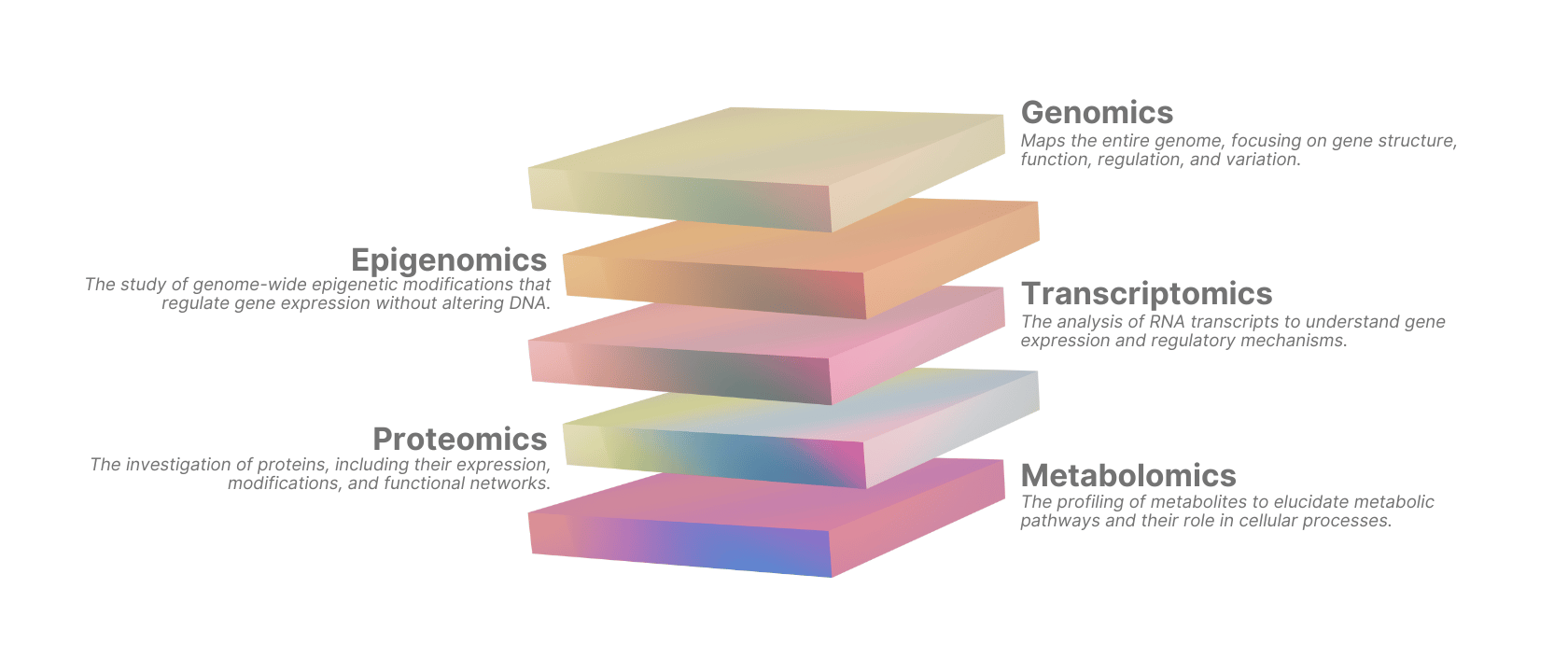
Figure 1: Convergent multi-omics evidence ranked at cell-state resolution for target prioritisation.
Working with the data
The single-cell transcriptomic layer usually defines the cell states against which the other layers are interpreted, and it is the layer most often returned from a bioinformatics queue with labels the experimental team cannot interrogate.
ScarfWeb is built for that layer: a browser-based, no-code environment for scRNA-seq analysis on Scarf, our memory-efficient engine published in Nature Communications. It is not a general multi-omic integration engine. Where a project needs chromatin, surface protein, spatial, or proteomic integration, our team supports that work alongside the platform.
If your target identification work depends on getting the cell-state layer right before the rest of the evidence is assembled, that is the part we can help with.
Related articles
The biology in your ligand-receptor analysis, and how to read it
Ligand-receptor analysis is easy to run and easy to over-read. How to get cell-cell communication biology from the ranked table, with LIANA and ScarfWeb.
Read more →
Spatial Transcriptomics and scRNA-seq: A Complementary Pair
The current state of spatial transcriptomics and scRNA-seq: single-cell-resolution platforms, foundation models for integration, spatial multi-omics, and 3D tissue atlases.
Read more →
Integrating Spatial Transcriptomics with Single-cell RNA-seq
How to integrate spatial transcriptomics with scRNA-seq: when spatial context helps, the key integration methods, and worked tissue examples.
Read more →